

Introduction
Protocols such as immunostaining on a chip are frequently searched for, at the moment. Cell fixation and immunostaining are critical techniques in cell biology and biomedical research. Cell fixation is the process of immobilising cells in a particular state, preserving their morphology, and preventing any further changes. This is crucial for the study of cellular structures, as it allows researchers to capture a “snapshot” of the cells at a specific moment in time. Fixation also inactivates cellular enzymes, preventing further biochemical processes, and maintaining the integrity of cellular components. This is important for preventing degradation and maintaining the stability of the cell for subsequent analysis. Fixed cells can be handled and stored for longer periods, allowing flexibility in post-processing and analysis.
Immunostaining is a technique that uses antibodies to detect and visualize specific proteins within cells. This is crucial for identifying the presence, location, and abundance of target proteins within the cellular context. Moreover, immunostaining provides spatial information by revealing where proteins are located within the cell. This allows for the analysis of images acquired under the microscope and the quantification of morphological, orientation, intensity and distance parameters.
This technical note provides the step-by-step procedure for fixing and labelling intestinal cells (HT29-MTX and Caco2) cultured on the upper fluidic channel of the Be-DoubleFlow device (Figure 1). The structures labelled were mucin 2 and zonula occludens.
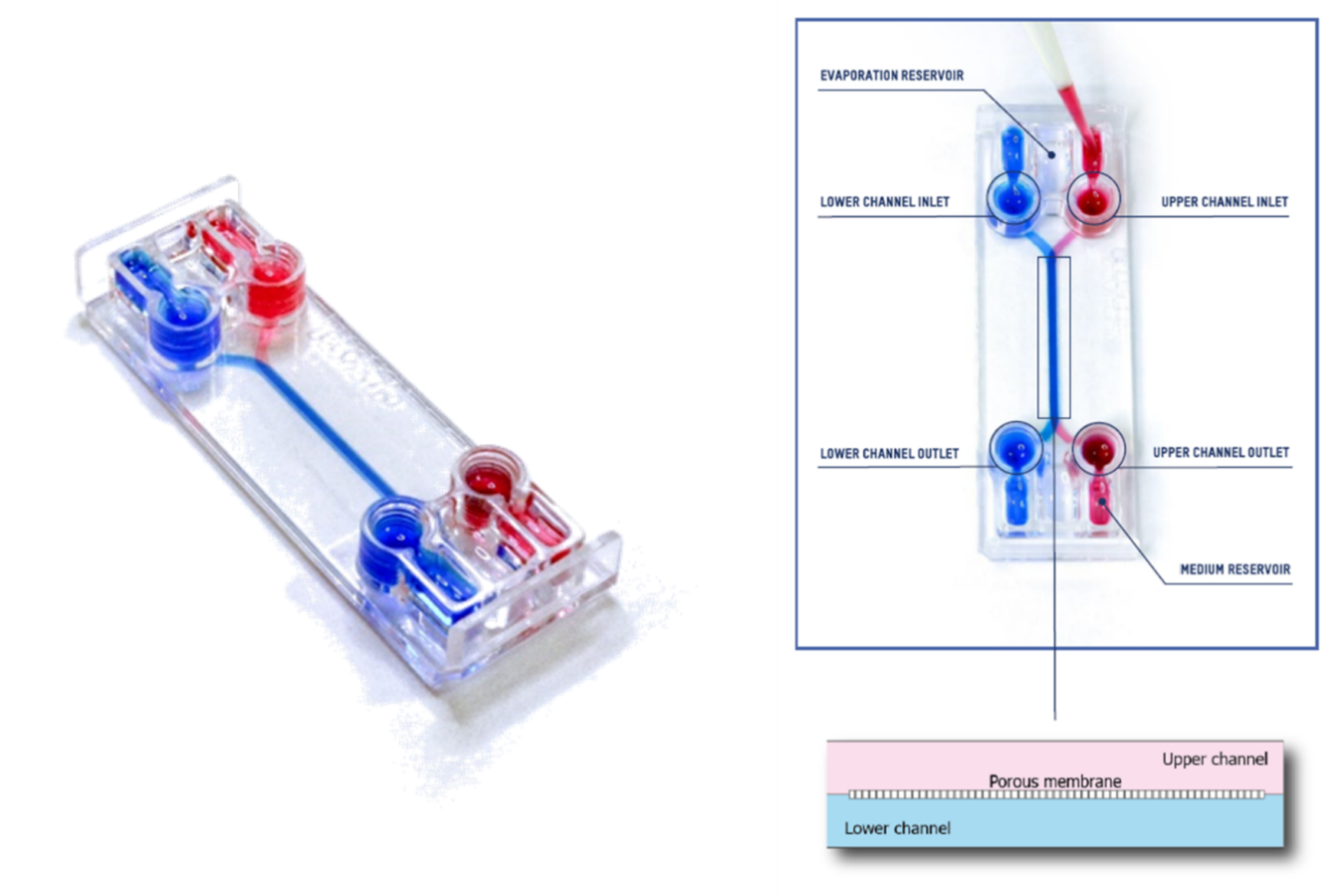
Figure 1. Be-DoubleFlow device.
Methodology
At the end of the experiment (Be-Doubleflow application notes: Gut-on-a-chip 1), cells were fixed with 4% paraformaldehyde (PFA).
Paraformaldehyde fixes and preserves cellular structures, including proteins, lipids, nucleic acids, and other components, maintaining their morphology and organization in the sample. It is a common choice in fluorescence microscopy due to its ability to preserve the fluorescence of proteins and fluorophores, enabling the detection of markers.
The following steps were followed in detail:
1. Remove the culture medium from the medium reservoirs and inlet/outlet wells, leaving the medium in the channel.
2. Add plenty of PBS (300 µL) and gently rock the chip manually to dilute the medium in the channel and perform gentle washes.
3. Carefully remove the PBS from the reservoirs and inlet/outlet wells and repeat this washing process as many times as necessary.
4. Add 100 µL PFA to the inlet well and tilt the chip to allow PFA to flow by gravity to the outlet well. Aspirate 100 µL from the outlet well. Repeat the process once more.
This will displace the residual PBS volume in the channel, allowing the permeabilizing solution to enter without completely emptying the channel.
It is possible to aspirate all the remaining PBS from inside the channel to empty it, and introduce 50 µL of PFA. (The channel’s volume will vary depending on the channel’s dimensions). When using this option, it is essential to ensure that the channel is entirely emptied to prevent overpressure and the introduction of air bubbles when refilling it. While it is entirely feasible to execute this step, we advise against doing it repeatedly in order to preserve the integrity of the culture within the channel.
5. Incubate for 15-20 minutes at room temperature.
6. Remove excess PFA from the input and output wells, leaving the channel filled.
7. Similar to step 2, add PBS and gently rock the chip to achieve a gentle washing effect. Remove the PBS and repeat this process three times to ensure thorough washing.
Permeabilization Process
The permeabilization process of the sample is essential for antibodies to be able to bind to internal cell structures. To achieve this step, non-ionic detergents are used, creating pores in the cell membrane and allowing the passage of substances and markers. In this instance, permeabilization was carried out using 0.1% Triton X-100 (Sigma Aldrich T9287-100ML) in PBS, followed by a rinse with 0.05% Tween 20 in PBS.
8. Add 100 µL of Triton to the inlet well and tilt the chip to allow Triton to flow by gravity to the outlet well. Aspirate 100 µL from the outlet well. Repeat the process once more.
This will displace the residual PBS volume in the channel, allowing the permeabilizing solution to enter without completely emptying the channel.
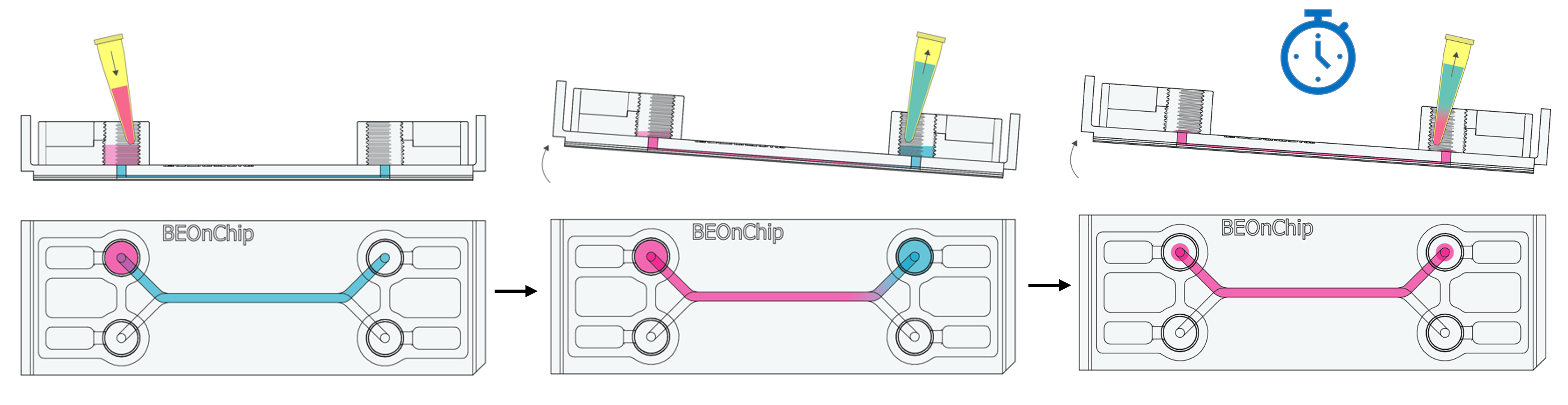
9. Incubate for 10 minutes at room temperature.
10. Remove excess Triton from the inlet and outlet wells, leaving the channel filled.
11. Perform the washing by adding a generous amount of Tween. Gentle rocking aids in achieving an effective wash. Remove the Tween and repeat this process three times to ensure thorough washing.

Blocking of the sample
The blocking of the biological sample in immunohistochemistry is crucial to ensure result specificity and reliability by minimizing the undesired binding of antibodies to other cellular structures. This reduces noise and enables a more precise and sensitive detection of the target of interest. To block non-specific interactions, samples were incubated with 5% Bovine Serum Albumin (A9418).
12. Add 100 µL of BSA to the inlet well and tilt the chip to allow BSA to flow by gravity to the outlet well. Aspirate 100 µL from the outlet well. Repeat the process once more.
This will displace the residual Tween volume in the channel, allowing the permeabilizing solution to enter without completely emptying the channel.

13. Incubate for 2 hours at room temperature.
Optimize the incubation time based on the culture type. For 2D and 2.5D cultures, 2-4 hours are usually sufficient. For 3D cultures, longer incubation times may be necessary.
14. Remove the BSA by carefully aspirating it from the channels completely. Ensure that no liquid residue or bubbles remain that could hinder antibody injection.
Primary and secondary antibodies
To label mucin 2 (MUC2 mouse sc-515032) and zonula occludens (ZO-1 rat Invitrogen 402200), primary antibodies were individually diluted at a ratio of 1:100 and secondary antibodies Alexa Fluor 488 (Goat anti-mouse A11001 Life Technologies) and Alexa Fluor 488 anti-rat 1:500 in 0.5% BSA.
15. Inject the primary antibodies by pipetting through the pinhole to fill the channel with the necessary volume (Standard channel 50 µL) and incubate overnight at 4°C. 
16. Perform several washes with 0.5% BSA following the recommendations in step 2 and 3.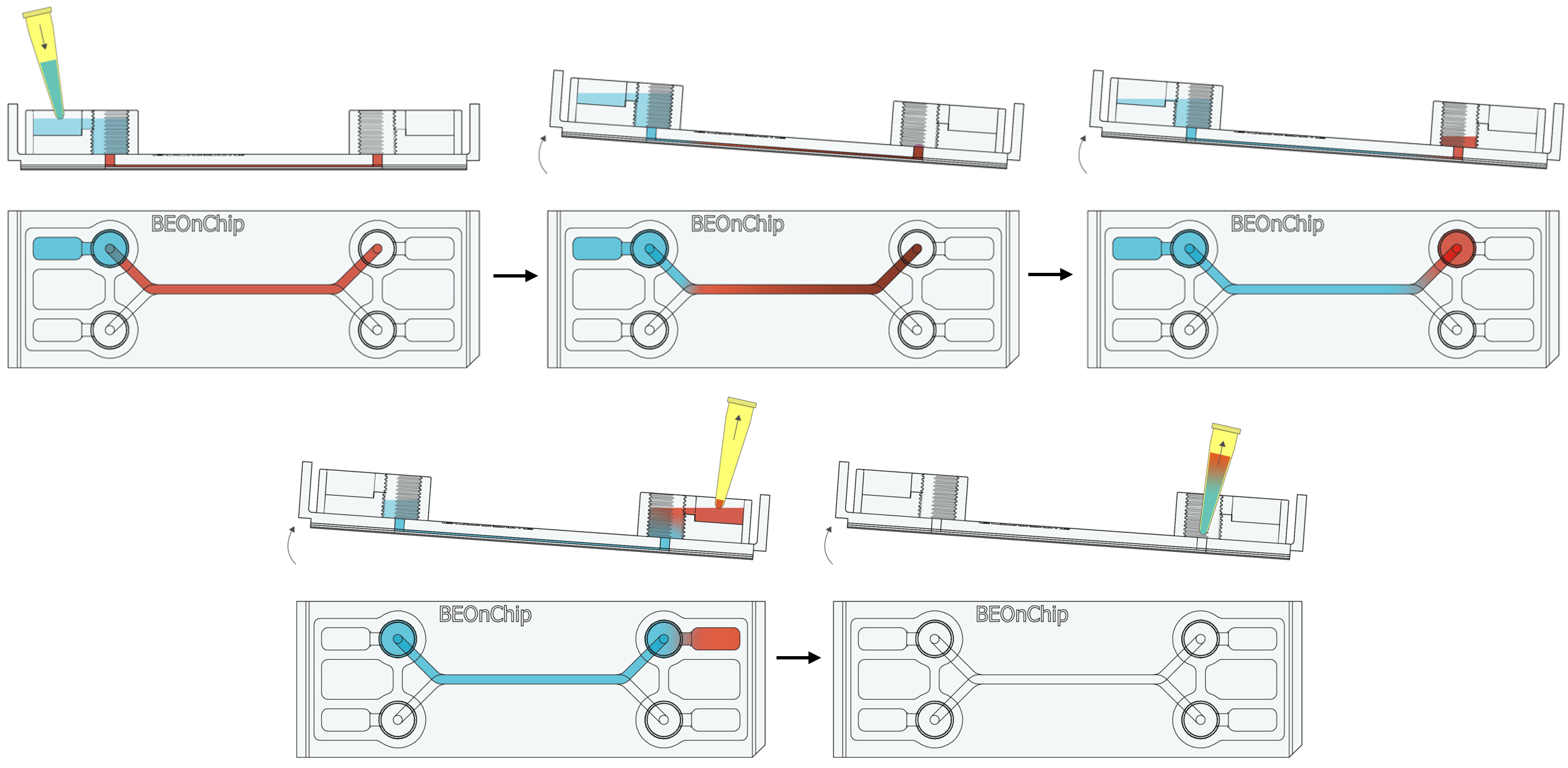 17. Completely empty the channel as in step 14 to introduce the secondary antibody through the pinhole and incubate for 2-3 hours at room temperature, protected from light.
17. Completely empty the channel as in step 14 to introduce the secondary antibody through the pinhole and incubate for 2-3 hours at room temperature, protected from light.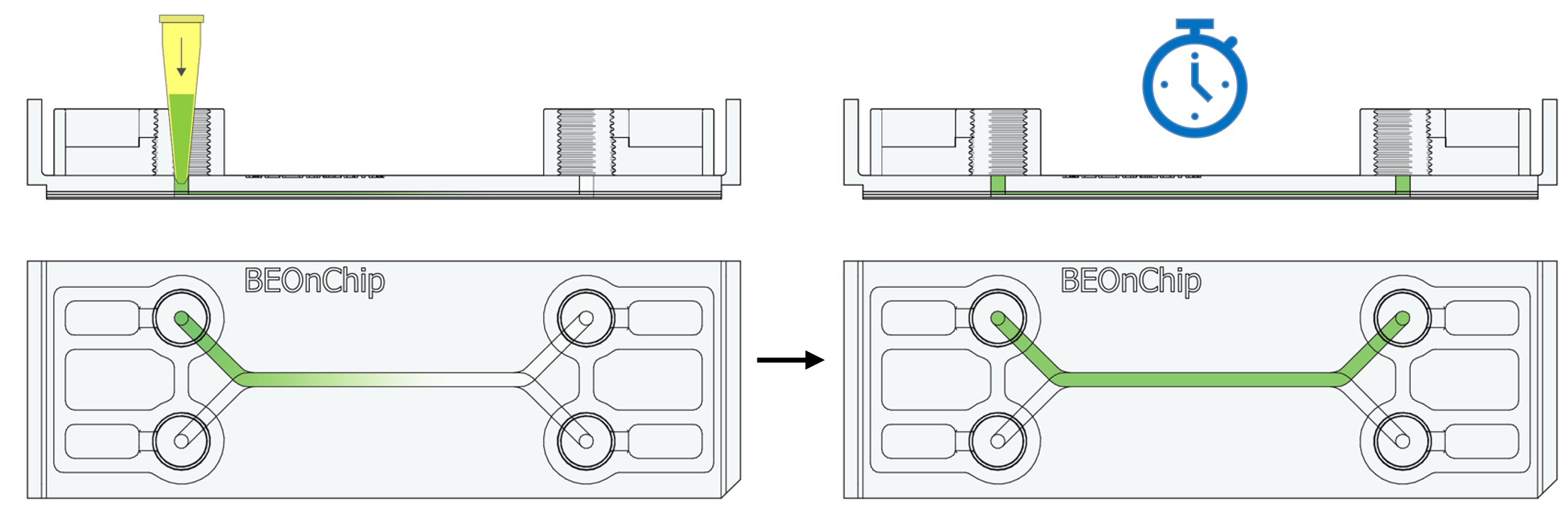 18. Wash with 0.5% BSA and then with PBS.
18. Wash with 0.5% BSA and then with PBS.
19. Additionally, incubate with DAPI (Sigma Aldrich D9542, 1:1000) for one hour at room temperature to stain the nuclei.
If possible, perform a negative control using the secondary antibody only (without primary antibody), especially with markers like MUC2, to distinguish background noise from actual mucin expression.
Finally, images were captured using a Leica Thunder microscope with a 20X objective.
Results and discussion
As depicted in figure 2, there is an evident expression of both mucin 2 and zonula occludens within the cultivated microfluidic device, confirming the proper formation of the epithelial barrier.

Figure 2. Fluorescence microscopy images of HT29MTX/Caco2 in a microfluidic device. Immunohistochemistry for MUC2 and ZO-1. Merge of ZO-1 with nuclei marked with DAPI. Scale bar: 100 µm.
Mucin 2
MUC2 is a vital secreted glycoprotein involved in the formation of the protective mucous layer that coats the intestinal epithelium, providing protection and lubrication. Its expression is crucial for faithfully replicating human intestinal characteristics, making it indispensable for studies aiming to mimic the physiological environment [1].
Tight junctions
Zonula occludens, also known as tight junctions, are cytoskeletal linker proteins that interact with peripheral cytoplasmic membrane proteins, including occludin and claudin, to establish robust cross-links with the membrane’s cytoskeleton, comprised of F-actin and myosin. Alongside intracellular signalling proteins, tight junction proteins orchestrate numerous cellular processes to preserve barrier integrity and regulate permeability. The presence of ZO-1 in an in vitro culture assists in mimicking barrier function and researching its regulation. For instance, studies can be conducted to assess how different compounds, such as drugs or nutrients, impact intestinal permeability and barrier integrity [2].
Immune responses and intestinal diseases
Furthermore, the intestinal barrier and mucus layer play a pivotal role in regulating the immune response and interaction with the intestinal microbiota. The expression of MUC2 and ZO-1 in an in vitro culture system offers a means to investigate how alterations in these structures impact the immune response and the composition of the microbiota[3,4].
On the other hand, the ability to sustain the expression of MUC2 and ZO-1 in an in vitro culture enables the study of intestinal diseases and disorders such as Crohn’s disease, ulcerative colitis, and other gastrointestinal conditions. This offers a suitable testing environment for assessing potential therapies and gaining a deeper understanding of the underlying mechanisms of these conditions [5].
These results represent the significant advancement in the control of culture conditions achieved by the incorporation of microfluidic devices in in vitro intestinal models. These devices allow enhanced precision in liquid flows and mechanical forces, simplifying the replication of specific physiological conditions within the intestine [6]. Other studies have reported that shear stress stimulates mucus production and enhances the expression of tight junction proteins in microfluidic devices [7].
Conclusions
In Beonchip’s microfluidic devices, it is possible to conduct protocols of in situ fixation and immunostaining in a chip, followed by visualization using fluorescence microscopy. Furthermore, in the specific application for the gut-on-chip model, the expression of mucin 2 and zonula occludens serves as indicators of barrier integrity.
Even though the results of this technical note were performed on the Be-Doubleflow device, the protocol can be replicated in all Beonchip devices.
Bibliography
- L. Sardelli, D. P. Pacheco, A. Ziccarelli, M. Tunesi, O. Caspani, A. Fusari, F. Briatico Vangosa, C. Giordano and P. Petrini, RSC Adv., 2019, 9, 15887–15899.
- C. Chelakkot, J. Ghim and S. H. Ryu, Exp. Mol. Med., 2018, 50, 103.
- T. Paradis, H. Bègue, L. Basmaciyan, F. Dalle and F. Bon, Int. J. Mol. Sci., 2021, 22, 1–21.
- M. Calvigioni, A. Panattoni, F. Biagini, L. Donati, D. Mazzantini, M. Massimino, C. Daddi, F. Celandroni, G. Vozzi and E. Ghelardi, Microbiol. Spectr., 2023, 11, e00336-23.
- S. S. Ghosh, J. Wang, P. J. Yannie and S. Ghosh, J. Endocr. Soc., 2020, 4, bvz039.
- A. Sontheimer-Phelps, D. B. Chou, A. Tovaglieri, T. C. Ferrante, T. Duckworth, C. Fadel, V. Frismantas, A. D. Sutherland, S. Jalili-Firoozinezhad, M. Kasendra, E. Stas, J. C. Weaver, C. A. Richmond, O. Levy, R. Prantil-Baun, D. T. Breault and D. E. Ingber, Cmgh, 2020, 9, 507–526.
- M. Chi, B. Yi, S. Oh, D. J. Park, J. H. Sung and S. Park, Biomed. Microdevices, 2015, 17, 1–10.
Author: Sandra González (ORCID).